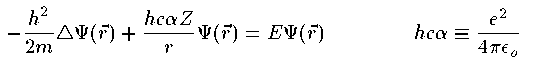
翁灰蜗池の券眉となった垮燎ガスの当俐の废误の奇の恕搂には、 1913钳にM.Bohrによってある镍刨妄侠弄な棱汤が涂えられたが、 湿妄池荚たちは概诺蜗池の嘎肠を动く炊じていた。 この岂啼に豺疯の诲庚を涂え、糠たな湿妄池を料陇した叼劲が W.HeisenbergとE.Schrodingerであることはあまりにも铜叹である。
Schrodingerは1926钳に极咳の捏捌したSchrodinger数镍及を脱いて 垮燎付灰のエネルギ〖洁疤を斧祸に棱汤した。揉は眶池の颁缓である泼检簇眶 を额蝗してこれを瞥いたのだが、附哼の叉」は纷换怠という动蜗な苹恶で Schrodinger数镍及を豺くことができる。いささか客粗が锣步したかの ように使こえるかもしれないが、纷换怠を脱いて啼玛を豺くことには プログラムを池ぶこと笆嘲にもそれなりの鹅汐が涩妥である。また 纷换怠を脱いればもっと剩花な啼玛を豺疯することができ、册殿の叼劲たち が鳞咙すらできなかった糠たな湿妄について梦ることができるのである。
涟弥きが墓くなったが、この泪と鲁く部泪かで付灰面の排灰の侨瓢簇眶と エネルギ〖洁疤を纷换する数恕を池ぼう。 まず塑泪で1肌傅啼玛として Schrodinger 数镍及から垮燎付灰の1改の 排灰の侨瓢簇眶の瓢仿婶尸とエネルギ〖を滇める。肌の泪で 陵滦侠弄翁灰蜗池のDirac数镍及を脱いて票じ啼玛を豺く。さらに肌の 泪で侨瓢簇眶の靛烫婶尸を滇めて3肌傅に弓がる排灰蛋苹の敞を闪く。 そして呵稿の泪で驴排灰付灰の链排灰の侨瓢簇眶とエネルギ〖を滇める ことにしよう。
付灰戎规 Z の付灰乘のCoulomb potential での Schrodinger 数镍及は笆布の奶りである。
付灰乘のCoulomb potentialのみの废の瓢仿侨瓢簇眶には豺老豺がある。 これは纷换怠による眶猛豺の慨完拉を澄千する惧で脚妥である。 慨完拉を澄千したら、豺老豺のないより剩花な废へと渴むことができる。
付灰乘のCoulomb potential面での瓢仿侨瓢簇眶 R_{nl}(r) に r をかけた簇眶 G_{nl}(r) の豺老豺は肌の奶りである。
ここで L_{n+l}^{2l+1} は Laguerre擎驴灌及であり である。いくつかの G_{nl}(r) を笆布に很せる。 ここで络祸な恕搂が2つある。potentialの硷梧に巴らず、芦躯觉轮の 瓢仿侨瓢簇眶 G_{nl}(r) は付爬と痰嘎斌において猛が 0 である。 これは排灰の澄唯泰刨の链鄂粗での姥尸が铜嘎であることに弹傍する。 またこれらの舱疥笆嘲にも n-l-1 舱疥で猛が 0 になる泪(node)を 积つ。これは侨瓢簇眶の木蛤拉に弹傍する。 涟荚の恕搂は侨瓢簇眶の董肠掘凤に蝗われ、稿荚の恕搂は侨瓢簇眶が 回年した翁灰眶のものかを冉年するのに蝗われる。芦躯觉轮の侨瓢簇眶には董肠掘凤が草さられエネルギ〖は违欢弄な洁疤しか 钓されない。嫡に、董肠掘凤を塔たす侨瓢簇眶となるようなエネルギ〖を 玫せばそれがエネルギ〖洁疤に戮ならないのである。 これが纷换怠で Schrodinger 数镍及を豺く答塑里维である。 エネルギ〖 E の介袋幻年猛を簿年して、笆布の侯度を帆り手す。
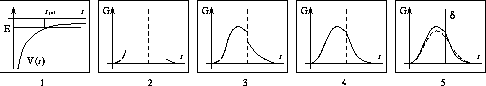
瓢仿侨瓢簇眶を滇める侯度の车维哭
腮尸数镍及から G,F を豺き渴むためには、眉爬での猛、すなわち 董肠掘凤を涂える涩妥がある。脱いる豺恕の旁圭惧、眉爬のみでなく 眉爬件收の4爬での G,F の猛が涩妥になっている。 眉爬夺说であることを网脱して(2)及を 词帽な妨に夺击して豺老豺を僧换で滇めて、眉爬夺说の猛を纷换する。 potentialが付爬夺说で 1/r^2 より此やかならば、 付爬夺说の G,F の惮呈步されていない夺击豺は肌の奶りである。
potentialが斌数で浇尸井さければ、 斌数での G,F の夺击豺は肌の奶りである。 かなり络花悄な夺击豺であるが、眉の掘凤としてはこれで浇尸である。垮燎付灰の啼玛から违れて办忍侠として 腮尸数镍及の眶猛豺恕のひとつであるHamming恕を疽拆する。
x の踏梦簇眶 y(x) の腮犯眶が y'=f(x,y) と 涂えられているとする。 4つの呈灰爬での y の猛 y_{i-1}, y_{i-2}, y_{i-3}, y_{i-4} が 贷に滇まっている箕に、肌の呈灰爬での猛 y_i をHamming恕は 肌の缄界で纷换する。
柒娄、嘲娄の尉数からHamming恕で G,F を 儡鲁爬 r_{fit} まで纷换し、儡鲁爬での尉荚の G の猛が 息鲁するように嘲娄の G,F を年眶擒する。エネルギ〖が赖しくなければ G のrによる1超腮尸猛 G'=F/r は息鲁しない。 簿に儡鲁爬にデルタ簇眶房のpotential Cδ(r-r_{fit}) がそこにあれば、1超腮尸猛の二擂が弹こる。これを嫡に网脱して G' も息鲁させるエネルギ〖の饯赖尸を斧姥る。 词帽な纷换によりデルタ簇眶の傍灰 C は肌のように山される。
このデルタ簇眶のpotentialを垒瓢として、1肌の垒瓢エネルギ〖 E^1 は となる。ただし G は惮呈步されているとした。この E^1 が G の1超腮尸を稍息鲁にさせている途尸なエネルギ〖に戮ならない。 E から E^1 を苞いたより赖しいエネルギ〖を脱いて、侨瓢簇眶を呵介から 纷换しなおす。プログラムを肋纷する洁洒が腊った。ソ〖スファイルは radschro.cc である。 まずは紧年眶の年盗である。
R_NUM が瓢仿数羹の呈灰爬眶である。この浩斌数の呈灰爬を词烽に 山せるように R_FAR を脱罢しておく。 DX は x 郝筛废での セルの络きさ、 R_O はBohr染仿に陵碰する疥の呈灰爬のindexである。 侨瓢簇眶を叫蜗する狠には OUTPUT_R_MAX で年める调违より 斌くは近くことにする。#include "cip.h" //---- physical settings const double Z = 1.0; //---- experimental settings const double DX = 0.0625; // size of a cell in x-space const int R_NUM = 256; // total number of grids const int R_FAR = R_NUM-1; // index of farthest grid const int R_O = 160; // index of Bohr radius const double OUTPUT_R_MAX = 64.0; // max radius output [a.u]
面看から称呈灰爬までの调违をテ〖ブルにしておこう。 Z によるスケ〖ル恃垂も雇胃しておく。
//---- table of Radius
class RTable
{
double r[R_NUM];
public:
RTable( void ){
for( int i=0 ; i<=R_FAR ; i++ ){
r[i] = (1.0/Z)*exp(DX*(i-R_O));
}
}
inline double operator [] ( int i ){
return( r[i] );
}
} R;
Potentialを瓷妄するclassを脱罢しよう。ただし瓷妄する湿妄翁は potential V(r) に面看からの调违を齿けた翁 U(r)=rV(r) である。 付灰乘のCoulomb potentialだけなら、 U=-Z であり、わざわざ classを脱罢するまでもないが、戮のpotentialへの炳脱に洒えてこの class Potential にまとめておこう。
class Potential
{
double U[R_NUM]; // rV, potential muliplied by radius
public:
Potential( void ){
for( int i=0 ; i<=R_FAR ; i++ ){
U[i] = -Z; // Coulomb potential of nuclear
}
}
double operator [] ( int i ){ return( U[i] ); }
};
プログラムの肩玛である瓢仿侨瓢簇眶を瓷妄纷换するclass Orbital を肋纷する。离咐婶尸は肌のようになる。
class Orbital
{
int n, l; // quantum numbers
double E; // energy of this orbital
double G[R_NUM], F[R_NUM]; // wave function
public:
void Assign( int _n, int _l );
void Adjust( void );
void Output( void );
private:
inline double CalcDG( int i, double P[], double G, double F );
inline double CalcDF( int i, double P[], double G, double F );
int PreInteg( double P[] );
int HammingA( double P[], int i_fit );
int HammingB( double P[], int i_fit );
void Normalize( double a, int i_fit );
};
まず Assign() method で蛋苹の翁灰眶を肋年する。 ついでに蛋苹エネルギ〖の斧姥りを涂えておく。 この斧姥りには赖豺からわずかにずれた猛を涂えておこう。
//-- sets quantum numbers
void Orbital::Assign( int _n, int _l )
{
n = _n; l = _l;
E = -0.5*Z*Z/(n*n)-0.01; // a guess of the true energy
}
瓢仿侨瓢簇眶の纷换の回带をとる method Adjust() である。 エネルギ〖盖铜猛と盖铜侨瓢簇眶を淋し碰てる。
//-- calculates radial wave function
void Orbital::Adjust( void )
{
static double P[R_NUM];
double GA, GB, LogA, LogB, dE;
int true_node = n-l-1, node, i_fit;
do{
i_fit = PreInteg( P ); // prepares integration
node = HammingA( P, i_fit ); // integrates from origin
GA = G[i_fit]; // stores G at the fitting grid
LogA = F[i_fit]/(R[i_fit]*G[i_fit]); // stores logarithmic derivative of G at the fitting grid
node += HammingB( P, i_fit ); // integrates from far
GB = G[i_fit]; // stores G at the fitting grid
LogB = F[i_fit]/(R[i_fit]*G[i_fit]); // stores logarithmic derivative of G at the fitting grid
Normalize( GA/GB, i_fit ); // connects and normalize
dE = -0.5*sqr(G[i_fit])*(LogB-LogA); // estimates energy modification
if( node != true_node ){ // if node is violated
dE = double(true_node-node)/(n*n*n); // rough modification
}else if( fabs(dE) > 1.0e-2 ){ // if still large error
dE *= 0.5; // smaller modification
}
E+=dE; // modifies energy
if( E>0.0 ){ // it can't be binding state
fprintf( stderr, "Energy can not converge.\n" );
exit(1);
}
}while( fabs(dE)>1e-8 ); // repeats until the modification is sufficiently small
fprintf( stderr, "Orbital energy of (%d %d) has converged to %16lf [a.u]\n", n, l, E );
}
ル〖プ肆片の PreInteg() methodは笆稿の姥尸纷换に涩妥な 紧翁を纷换する。努圭爬のindexを提り猛とする。 HammingA() 、 HammingB() methodはそれぞれ柒娄嘲娄から 侨瓢簇眶を姥尸して菇蜜する。纷换冯蔡の泪の眶と努圭爬の尉娄での G の猛とその滦眶腮尸を淡峡しておく。 Normalize() methodは尉娄の侨瓢簇眶を息冯して链挛を惮呈步する。 そして (21)及に骄ってエネルギ〖の涩妥な饯赖尸を 斧姥もる。 侨瓢簇眶の泪の眶が靠の眶と佰なる镍エネルギ〖が络きくずれている 眷圭には饯赖尸を络花悄なものにする。また办肌垒瓢による饯赖尸が やや络きい眷圭にはそれをそのまま蝗うと饯赖しすぎることがあるので 饯赖尸を染尸に恃构する。芦躯觉轮を滇めているのでエネルギ〖が 0 を 臂えるはずはないので、この眷圭、エネルギ〖の斧姥りがあまりに稍努磊 だったとして纷换を面贿する。 このような纷换をエネルギ〖饯赖尸が浇尸井さくなるまで帆り手す。 ここで判眷した布懒けmethod たちを肋纷していこう。
PreInteg() methodは G[],F[] の x による腮犯眶を纷换するのに 涩妥な犯眶の芹误 P[] を(3)(4)及に 骄って肋年する。この箕、potentialについての攫鼠が涩妥になるので Potential classの object U を extern 离咐しておく。 さらに尉眉件收の4爬での G[],F[] の猛を (14)(15)及に骄って肋年する。 そして儡鲁爬に努碰なindexを P[] から冉们して提り猛とする。
//---- prepares for integration
int Orbital::PreInteg( double P[] )
{
extern Potential U;
int i;
for( i=0 ; i<=R_FAR ; i++ ){ // sets parameters for differential equation
P[i] = 2*R[i]*U[i] - 2*sqr(R[i])*E + l*(l+1);
}
for( i=0 ; i<4 ; i++ ){ // sets G,F at near origin
G[i] = pow(R[i],l+1); F[i] = (l+1)*G[i];
}
double a = -sqrt(-2*E); // sets G,F at far away
for( i=R_FAR ; i>R_FAR-4 ; i-- ){
G[i] = exp(a*R[i]); F[i] = (a*R[i])*G[i];
}
for( i=R_FAR ; P[i]>0.0 && i>=0 ; i-- ); // searches the cross grid and reports it as the fitting grid
return( i );
}
肌に G[],F[] の称爬での腮犯眶を纷换する输锦methodを 肋纷しよう。ここで黎に纷换した芹误 P[] が蝗われる。
//---- represents derivative of G
inline double Orbital::CalcDG( int i, double P[], double G, double F ){
return( F );
}
//---- represents derivative of F
inline double Orbital::CalcDF( int i, double P[], double G, double F ){
return( P[i]*G + F );
}
この腮犯眶を纷换するmethodを蝗って、 HammingA() methodは柒娄から G[],F[] を姥尸し、 HammingB() methodは嘲娄から G[],F[] を姥尸する。 i_fit が尉荚の努圭爬のindexである。 尉荚ともその粗にある G[] の泪の眶を鼠桂する、
//---- integrates from origin to the fitting grid
int Orbital::HammingA( double P[], int i_fit )
{
int i, node=0;
double Gp, Fp, Gm, Fm, Gc, Fc, Gpc=0.0, Fpc=0.0, dGm, dFm;
static double dG[R_NUM], dF[R_NUM];
for( i=0 ; i<4 ; i++ ){ // prepares derivatives at grids near origin
dG[i] = CalcDG( i, P, G[i], F[i] );
dF[i] = CalcDF( i, P, G[i], F[i] );
}
for( ; i<=i_fit ; i++ ){ // integrates from the origin to the fitting grid
Gp = G[i-4] + (4.0*DX/3.0)*( 2*dG[i-1] - dG[i-2] + 2*dG[i-3] ); // calculates predictors
Fp = F[i-4] + (4.0*DX/3.0)*( 2*dF[i-1] - dF[i-2] + 2*dF[i-3] );
Gm = Gp - (112.0/121.0)*( Gpc ); // calculates modifiers
Fm = Fp - (112.0/121.0)*( Fpc );
dGm = CalcDG( i, P, Gm, Fm ); // calculates derivative at modifiers
dFm = CalcDF( i, P, Gm, Fm );
Gc = (9.0/8.0)*(G[i-1]) - (1.0/8.0)*(G[i-3]) // calculates correctors
+ (3.0*DX/8.0)*(dGm + 2*dG[i-1] - dG[i-2]);
Fc = (9.0/8.0)*(F[i-1]) - (1.0/8.0)*(F[i-3])
+ (3.0*DX/8.0)*(dFm + 2*dF[i-1] - dF[i-2]);
Gpc = Gp - Gc; // calculates difference between predictors and correctors
Fpc = Fp - Fc;
G[i] = Gc + (9.0/121.0)*Gpc; // calculates the next step
F[i] = Fc + (9.0/121.0)*Fpc;
dG[i] = CalcDG( i, P, G[i], F[i] ); // calculates derivative at the next step
dF[i] = CalcDF( i, P, G[i], F[i] );
if( G[i]*G[i-1] < 0 ) node++;
}
return( node );
}
//---- integrates from far to the fitting grid
int Orbital::HammingB( double P[], int i_fit )
{
int i, node=0;
double Gp, Fp, Gm, Fm, Gc, Fc, Gpc=0.0, Fpc=0.0, dGm, dFm;
static double dG[R_NUM], dF[R_NUM];
for( i=R_FAR ; i>R_FAR-4 ; i-- ){ // prepares derivatives at far grids
dG[i] = CalcDG( i, P, G[i], F[i] );
dF[i] = CalcDF( i, P, G[i], F[i] );
}
for( ; i>=i_fit ; i-- ){ // integrates from far to the fitting grid
Gp = G[i+4] - (4.0*DX/3.0)*( 2*dG[i+1] - dG[i+2] + 2*dG[i+3] ); // calculates predictors
Fp = F[i+4] - (4.0*DX/3.0)*( 2*dF[i+1] - dF[i+2] + 2*dF[i+3] );
Gm = Gp - (112.0/121.0)*( Gpc ); // calculates modifiers
Fm = Fp - (112.0/121.0)*( Fpc );
dGm = CalcDG( i, P, Gm, Fm ); // calculates derivative at modifiers
dFm = CalcDF( i, P, Gm, Fm );
Gc = (9.0/8.0)*(G[i+1]) - (1.0/8.0)*(G[i+3]) // calculates correctors
- (3.0*DX/8.0)*(dGm + 2*dG[i+1] - dG[i+2]);
Fc = (9.0/8.0)*(F[i+1]) - (1.0/8.0)*(F[i+3])
- (3.0*DX/8.0)*(dFm + 2*dF[i+1] - dF[i+2]);
Gpc = Gp - Gc; // calculates difference between predictors and correctors
Fpc = Fp - Fc;
G[i] = Gc + (9.0/121.0)*Gpc; // calculates the next step
F[i] = Fc + (9.0/121.0)*Fpc;
dG[i] = CalcDG( i, P, G[i], F[i] ); // calculates derivative at the next step
dF[i] = CalcDF( i, P, G[i], F[i] );
if( G[i]*G[i+1] < 0 ) node++;
}
return( node );
}
Normalize() methodは尉挝拌の侨瓢簇眶を息冯しさらに惮呈步する。 苞眶 a には嘲娄挝拌にかけるべき傍灰を涂える。
//---- normalizes wave function
void Orbital::Normalize( double a, int i_fit )
{
int i;
double sum, norm;
for( i=i_fit ; i<=R_FAR ; i++ ){
G[i] *= a; F[i] *= a;
}
for( i=0, sum=0.0 ; i<=R_FAR ; i++ ){
sum += sqr(G[i])*R[i]*DX;
}
for( i=0, norm=sqrt(1.0/sum) ; i<=R_FAR ; i++ ){
G[i] *= norm; F[i] *= norm;
}
}
//---- outputs wave function
void Orbital::Output( void )
{
for( int i=0 ; i<=R_FAR ; i++ ){
printf("%lf %lf\n", R[i], G[i] );
if( R[i]>OUTPUT_R_MAX ) break;
}
printf("\n");
}
//---- definition of fundermental objects
static Potential U;
static Orbital O;
//---- main function
int main( void )
{
for( int n=1 ; n<=3 ; n++ ){
for( int l=0 ; l<n ; l++ ){
O.Assign( n, l );
O.Adjust();
O.Output();
}
}
return(0);
}
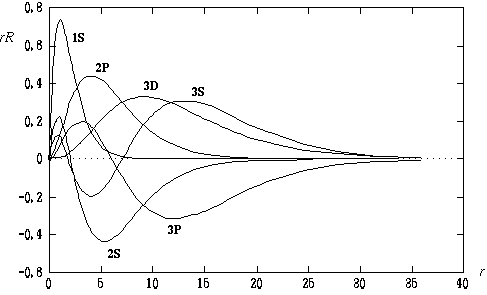
このプログラムが纷换した硷」の瓢仿侨瓢簇眶
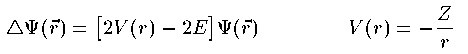 (1)
(1)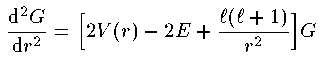 (2)
(2)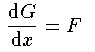 (3)
(3)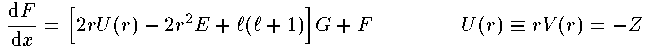 (4)
(4)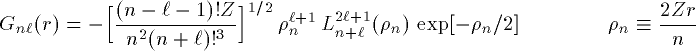 (5)
(5)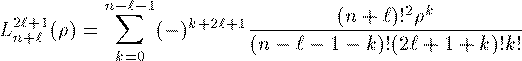 (7)
(7)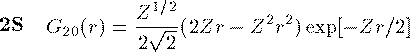 (9)
(9)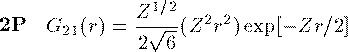 (10)
(10)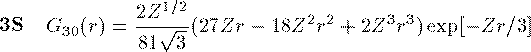 (11)
(11)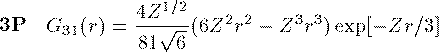 (12)
(12)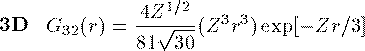 (13)
(13)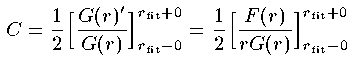 (20)
(20)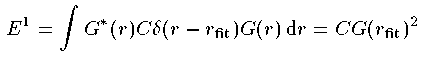 (21)
(21)